How can so-called first principle calculations be used more simply and efficiently than before to investigate materials with the aid of computers and develop new types of materials? Max Großmann is investigating this question in his doctoral thesis at the Institute of Physics at TU Ilmenau under the supervision of Prof. Erich Runge. Together with his colleague Malte Grunert, the Master's graduate in Technical Physics has developed a new method for so-called GW calculations, thus opening up possibilities for deciphering the secrets of matter at the atomic level more quickly in the future. The results of the investigations have just been published in the renowned Nature Journal NPJ Computational Materials:
Research
Tracking down the secrets of matter
Team of physicists develops new method for computer-aided materials research

Whether designing new materials for energy storage or medicines, predicting the optical properties of materials or detecting material damage, machine learning methods have recently become increasingly popular in materials science. Among other things, they can be used to investigate the optical and electronic structure, i.e. the arrangement of electrons in the material. The quantum mechanical interactions to be taken into account provide information about what happens when a material is energized or irradiated with light, among other things.
However, one of the biggest hurdles in this rapidly growing field of research is the lack of robust training data required for the efficient application of machine learning methods. While high-throughput experiments are ideal, they are often prohibitively expensive and labor-intensive. Therefore, materials scientists often resort to computer simulations, which, unlike empirical models without external parameters, work exclusively with analytical formulas. They are known as ab initio or first principle calculations.
The most widely used and so far most successful ab initio method for calculating the properties of molecules and solids is the so-called density functional theory (DFT). In recent decades, it has contributed to major advances in materials science and opened up new possibilities for the selection and design of novel materials for many practical applications - from solar cells, catalysts and batteries to high-performance electronics.
Automating calculations simply, robustly and efficiently
However, this method is slowly reaching its limits due to its shortcomings, such as the fact that it greatly underestimates the band gap of semiconductors and insulators. A promising alternative is the so-called GW method, which is now widely used in studies on the electronic structure and optical properties of materials, but is also very computationally intensive. This method was the focus of Max Großmann's investigations, which he carried out as part of his doctoral thesis together with Malte Grunert at the Institute of Physics under the supervision of Prof. Erich Runge. In an article entitled "A robust, simple, and efficient convergence workflow for GW calculations", which has just been published in the renowned Nature Journal NPJ Computational Materials , the young scientists explain how these calculations can be automated simply, robustly and efficiently.
"The focus of our investigations was the question: How can we improve the data from previous DFT calculations using the GW method with modern computing resources within a reasonable amount of time and effort?", explains Großmann: "It was important to find out how many GW calculations we can perform in a reasonable amount of time. Always with the long-term goal in mind of being able to use the data for machine learning in the future."
After the two scientists had initially implemented and tested a workflow published last year for performing GW calculations in their code, they asked themselves simple questions such as "Can this workflow be made more robust, simpler and more efficient?", "Does the algorithm have to be so complex?" and "Can we independently optimize the various convergence parameters that ensure that the simulation gets closer to the best possible result step by step?".
The right calculations in the right order
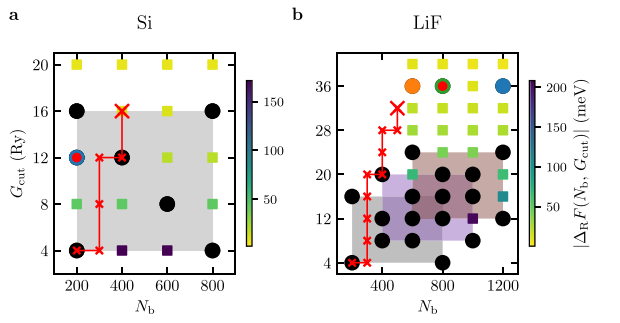
Großmann: "Following the motto 'the simplest model is often the best', we finally decided to develop our own robust, simple and efficient workflow and performed calculations for 70 semiconducting and insulating materials on the local computing cluster at the university computing center." The performance and accuracy of this GW workflow was compared to the previously published one, still using expensive reference calculations as a comparison. They also investigated whether the independence of certain convergence parameters could be exploited to further speed up the workflow.
The scientists describe the results of their study in detail in their paper: "With our GW workflow, which is easy to implement both automatically and manually, the calculation time can be reduced by more than a factor of two without loss of accuracy".
In addition, the scientists found that a further factor of 10 in computation time can be saved if the independence of the convergence parameters shown in the paper is used.
"If you integrate these findings into a workflow, you can significantly increase the computational efficiency of the GW calculation without having to change the underlying ab initio code by simply performing the right calculations in the right order," summarizes Großmann. "This makes the workflow ideal for future GW calculations with high data throughput and paves the way for the use of computer simulations that go beyond DFT, such as many-body perturbation theory in large-scale material screening projects." Thus, the new method can help to research new materials for various high-tech applications better, faster and more efficiently in the future.
Original publication
Großmann, M., Grunert, M. & Runge, E. A robust, simple, and efficient convergence workflow for GW calculations. npj Comput Mater 10, 135 (2024). https://doi.org/10.1038/s41524-024-01311-9